Author: Nick Semenkovich (semenko@alum.mit.edu)




![]()
![]()

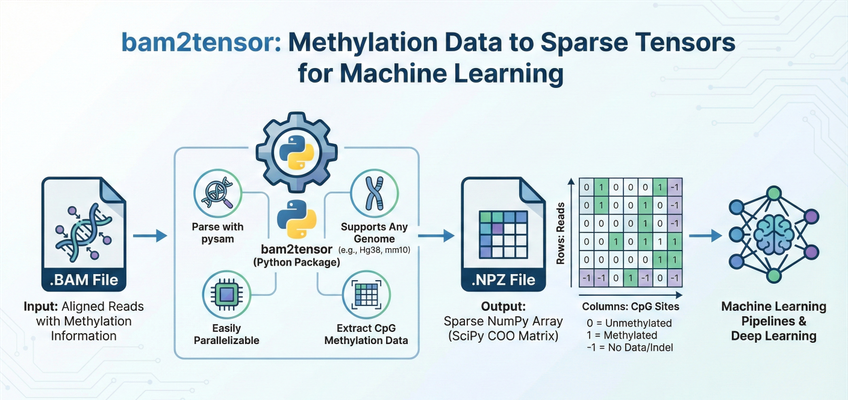
bam2tensor is a Python package for converting bisulfite-sequencing (BS-seq) and enzymatic methylation sequencing (EM-seq) .bam files to sparse tensor representations of DNA methylation data. It extracts read-level methylation states from CpG sites and outputs efficient sparse COO matrices as .npz files, ready for deep learning pipelines.
- Features
- Requirements
- Installation
- Quick Start
- Usage
- Inspecting Output Files
- Output Data Structure
- Supported Aligners
- Performance Tips
- API Reference
- Contributing
- AI Coding Agents
- License
- Credits
- BAM Parsing: Efficiently parses
.bamfiles using pysam - Complete CpG Extraction: Extracts methylation data from all CpG sites genome-wide
- Multi-Genome Support: Works with any reference genome (GRCh38/hg38, T2T-CHM13, mm10, etc.)
- Sparse Storage: Stores data in sparse COO matrix format for memory-efficient loading
- NumPy/SciPy Integration: Exports to
.npzfiles compatible with NumPy and SciPy - Efficient Algorithm: Linear-scan algorithm ensures minimal memory usage with no read duplication
- Batch Processing: Process multiple BAM files with directory recursion
- Caching: CpG site indexing is cached to accelerate repeated runs on the same genome
- Quality Filtering: Configurable mapping quality thresholds
- Python 3.10 or higher
- A reference genome FASTA file (must match the genome used for alignment)
- Indexed BAM files (
.bamwith corresponding.bam.baiindex files)
Core dependencies are automatically installed:
pysam- BAM file handlingbiopython- FASTA parsingscipy- Sparse matrix operationsnumpy- Numerical operationsclick- Command-line interfacetqdm- Progress bars
pip install bam2tensorgit clone https://github.com/mcwdsi/bam2tensor.git
cd bam2tensor
pip install .git clone https://github.com/mcwdsi/bam2tensor.git
cd bam2tensor
uv sync# Basic usage with a single BAM file
bam2tensor \
--input-path sample.bam \
--reference-fasta GRCh38.fa \
--genome-name hg38
# This creates: sample.methylation.npzProcess a single bisulfite-sequencing or EM-seq BAM file:
bam2tensor \
--input-path /path/to/aligned_reads.bam \
--reference-fasta /path/to/reference.fa \
--genome-name hg38This will:
- Parse the reference FASTA to identify all CpG sites (cached for future runs)
- Extract methylation states from each read in the BAM file
- Output a sparse matrix to
aligned_reads.methylation.npz
Don't have a local reference FASTA? bam2tensor can download and cache common reference genomes automatically:
# Download hg38 and process a BAM file in one command
bam2tensor \
--input-path sample.bam \
--download-reference hg38
# List available genomes
bam2tensor --input-path sample.bam --list-genomesAvailable genomes: hg38, hg19, mm10, T2T-CHM13. Downloaded references are cached in ~/.cache/bam2tensor/ for future runs.
Process all BAM files in a directory recursively:
bam2tensor \
--input-path /path/to/bam_directory/ \
--reference-fasta /path/to/reference.fa \
--genome-name hg38 \
--verboseEach BAM file will generate a corresponding .methylation.npz file in the same location.
Write output files to a specific directory instead of next to the input BAMs:
bam2tensor \
--input-path /shared/bams/ \
--reference-fasta /shared/ref/GRCh38.fa \
--genome-name hg38 \
--output-dir /scratch/methylation_output/This is useful on HPC clusters where input directories may be read-only or on slow shared storage.
For non-human genomes or custom chromosome sets:
# Mouse genome (mm10)
bam2tensor \
--input-path mouse_sample.bam \
--reference-fasta mm10.fa \
--genome-name mm10 \
--expected-chromosomes "chr1,chr2,chr3,chr4,chr5,chr6,chr7,chr8,chr9,chr10,chr11,chr12,chr13,chr14,chr15,chr16,chr17,chr18,chr19,chrX,chrY"
# T2T-CHM13 human genome
bam2tensor \
--input-path sample.bam \
--reference-fasta chm13v2.0.fa \
--genome-name T2T-CHM13 \
--expected-chromosomes "chr1,chr2,chr3,chr4,chr5,chr6,chr7,chr8,chr9,chr10,chr11,chr12,chr13,chr14,chr15,chr16,chr17,chr18,chr19,chr20,chr21,chr22,chrX,chrY"Usage: bam2tensor [OPTIONS]
Extract read-level methylation data from an aligned bisulfite-seq or EM-seq
.bam file and export the data as a SciPy sparse matrix.
Options:
--version Show the version and exit.
--input-path PATH Input .bam file OR directory to recursively
process. [required]
--genome-name TEXT A custom string referring to your genome
name, used to save a cache file (e.g. hg38,
hg39-no-alt, etc.).
--expected-chromosomes TEXT A comma-separated list of chromosomes to
expect in the .fa genome. Defaults to hg38
chromosomes.
--reference-fasta FILE Reference genome fasta file (critical to
determine CpG sites).
--quality-limit INTEGER Quality filter for aligned reads (default =
20)
--verbose Verbose output.
--skip-cache De-novo generate CpG sites (slow).
--debug Debug mode (extensive validity checking +
debug messages).
--overwrite Overwrite output file if it exists.
--output-dir DIRECTORY Output directory for .methylation.npz files.
Defaults to same directory as the input BAM.
--download-reference [t2t-chm13|hg19|hg38|mm10]
Download and cache a known reference genome
(e.g. hg38, hg19, mm10, T2T-CHM13). Use
--list-genomes to see options.
--list-genomes List available reference genomes for
download and exit.
--help Show this message and exit.
| Option | Description |
|---|---|
--input-path |
Path to a single .bam file or a directory. If a directory is provided, all .bam files are processed recursively. |
--genome-name |
An identifier for your reference genome (e.g., hg38, mm10). Used to name the cache file for CpG site positions. |
--expected-chromosomes |
Comma-separated list of chromosome names to process. Chromosomes not in this list are skipped. Defaults to human autosomes + sex chromosomes. |
--reference-fasta |
Path to the reference genome FASTA file. Must match the genome used for alignment. |
--quality-limit |
Minimum mapping quality score (MAPQ) for reads to be included. Default is 20. |
--verbose |
Enable detailed progress output including per-chromosome progress bars. |
--skip-cache |
Force regeneration of CpG site cache. Useful if you've modified the reference or chromosome list. |
--debug |
Enable extensive validation and debug output. Slower but useful for troubleshooting. |
--overwrite |
Overwrite existing .methylation.npz files. Without this flag, existing outputs are skipped. |
--output-dir |
Write .methylation.npz files to this directory instead of next to the input BAMs. Created automatically if it doesn't exist. |
--download-reference |
Download and cache a known reference genome. Choices: hg38, hg19, mm10, T2T-CHM13. Replaces --reference-fasta. |
--list-genomes |
List available reference genomes for --download-reference and exit. |
Use bam2tensor-inspect to view a summary of any .methylation.npz file without writing Python:
$ bam2tensor-inspect sample.methylation.npz
sample.methylation.npz
Genome: hg38
Chromosomes: 24 (chr1, chr2, ... chrX, chrY)
Reads: 1,423,891
CpG sites: 28,217,448
Data points: 12,847,322 (sparsity: 99.97%)
CpG index CRC32: a1b2c3d4
bam2tensor: v2.3
File size: 14.2 MBYou can pass multiple files at once:
$ bam2tensor-inspect *.methylation.npzThis works on files produced by older versions of bam2tensor too (metadata fields will be omitted).
bam2tensor generates one .methylation.npz file per input BAM file. Each file contains a SciPy sparse COO matrix (scipy.sparse.coo_matrix) with the following structure:
| Dimension | Represents |
|---|---|
| Rows | Unique sequencing reads (primary alignments that pass quality and flag filters, numbered sequentially as encountered across chromosomes) |
| Columns | CpG sites from the reference genome, ordered by genomic position across all chromosomes (chr1, chr2, ..., chrX, chrY). Column i maps to the i-th CpG dinucleotide in the reference FASTA. |
The column dimension is determined entirely by the reference genome: it equals the total number of CpG sites across all --expected-chromosomes. For example, hg38 with default chromosomes has ~28 million CpG columns. To map column indices back to genomic coordinates (e.g., column 12345 → chr1:29503), use the GenomeMethylationEmbedding class with the same reference FASTA and chromosome list (see Working with Genomic Coordinates below).
| Value | Meaning |
|---|---|
1 |
Methylated (cytosine preserved as C after bisulfite/enzymatic conversion) |
0 |
Unmethylated (cytosine converted to T by bisulfite/enzymatic treatment) |
-1 |
No data (indel, SNV, or other non-C/T base at a CpG position) |
Note: The matrix uses SciPy's COO sparse format, which explicitly stores all non-zero values. Unmethylated sites (value 0) are stored as explicit entries. Positions not covered by a read are simply absent from the matrix (implicit zero, which is distinct from the explicit 0 = unmethylated).
Each .methylation.npz file includes a metadata.json entry inside the ZIP archive with provenance information:
| Field | Description |
|---|---|
bam2tensor_version |
Version of bam2tensor that produced the file |
genome_name |
Genome identifier (e.g., hg38, mm10) |
expected_chromosomes |
List of chromosomes included in the column mapping |
total_cpg_sites |
Total number of CpG columns in the matrix |
cpg_index_crc32 |
CRC32 checksum of the CpG site positions (verifies identical column semantics) |
This metadata is ignored by scipy.sparse.load_npz, so existing code continues to work. To read it:
from bam2tensor.metadata import read_npz_metadata
meta = read_npz_metadata("sample.methylation.npz")
if meta is not None:
print(f"Genome: {meta['genome_name']}")
print(f"CpG sites: {meta['total_cpg_sites']:,}")
print(f"CpG index CRC32: {meta['cpg_index_crc32']}")The cpg_index_crc32 field uniquely identifies the column mapping. Two files with the same CRC32 have identical column semantics (same chromosomes, same CpG positions, same order) and their matrices can be directly stacked or compared. The metadata is also accessible without bam2tensor installed, since .npz files are ZIP archives:
unzip -p sample.methylation.npz metadata.json | python -m json.toolimport scipy.sparse
import numpy as np
# Load the sparse matrix
methylation_matrix = scipy.sparse.load_npz("sample.methylation.npz")
print(f"Matrix shape: {methylation_matrix.shape}")
print(f"Number of reads: {methylation_matrix.shape[0]}")
print(f"Number of CpG sites: {methylation_matrix.shape[1]}")
print(f"Non-zero entries: {methylation_matrix.nnz}")
print(f"Sparsity: {1 - methylation_matrix.nnz / np.prod(methylation_matrix.shape):.4%}")For small regions or when dense operations are needed:
# Convert entire matrix to dense (warning: may use significant memory)
dense_matrix = methylation_matrix.toarray()
# Convert to CSR format for efficient row slicing
csr_matrix = methylation_matrix.tocsr()
# Get methylation data for reads 0-99
subset = csr_matrix[0:100, :].toarray()
# Convert to CSC format for efficient column slicing
csc_matrix = methylation_matrix.tocsc()
# Get data for CpG sites 1000-1099
cpg_subset = csc_matrix[:, 1000:1100].toarray()To map between matrix column indices and genomic coordinates, use the GenomeMethylationEmbedding class:
from bam2tensor.embedding import GenomeMethylationEmbedding
# Load or recreate the embedding used during extraction
embedding = GenomeMethylationEmbedding(
genome_name="hg38",
expected_chromosomes=["chr" + str(i) for i in range(1, 23)] + ["chrX", "chrY"],
fasta_source="/path/to/GRCh38.fa",
)
# Convert matrix column index to genomic position
chrom, pos = embedding.embedding_to_genomic_position(12345)
print(f"Column 12345 corresponds to {chrom}:{pos}")
# Convert genomic position to matrix column index
col_idx = embedding.genomic_position_to_embedding("chr1", 10525)
print(f"chr1:10525 is at column {col_idx}")
# Get total number of CpG sites
print(f"Total CpG sites: {embedding.total_cpg_sites:,}")import scipy.sparse
import numpy as np
# Load the data
matrix = scipy.sparse.load_npz("sample.methylation.npz")
csr = matrix.tocsr()
# Calculate per-CpG methylation rates (excluding -1 values)
methylation_rates = []
for cpg_idx in range(matrix.shape[1]):
col_data = csr.getcol(cpg_idx).toarray().flatten()
# Filter out -1 (no data) and positions with no coverage
valid_data = col_data[(col_data >= 0)]
if len(valid_data) > 0:
rate = np.mean(valid_data)
else:
rate = np.nan
methylation_rates.append(rate)
methylation_rates = np.array(methylation_rates)
print(f"Mean methylation rate: {np.nanmean(methylation_rates):.2%}")
print(f"CpG sites with coverage: {np.sum(~np.isnan(methylation_rates)):,}")import torch
import scipy.sparse
import numpy as np
# Load sparse matrix
matrix = scipy.sparse.load_npz("sample.methylation.npz")
# Convert to PyTorch sparse tensor
coo = matrix.tocoo()
indices = torch.LongTensor(np.vstack((coo.row, coo.col)))
values = torch.FloatTensor(coo.data)
shape = torch.Size(coo.shape)
sparse_tensor = torch.sparse_coo_tensor(indices, values, shape)
print(f"PyTorch sparse tensor shape: {sparse_tensor.shape}")
# For models that need dense input (specific region)
region_start, region_end = 0, 1000
dense_region = matrix.tocsc()[:, region_start:region_end].toarray()
dense_tensor = torch.FloatTensor(dense_region)bam2tensor supports BAM files from bisulfite-aware and EM-seq-aware aligners that include strand information tags:
| Aligner | Tag | Values |
|---|---|---|
| Bismark | XM |
Z (methylated CpG), z (unmethylated CpG) |
| Biscuit | YD |
f (forward/OT/CTOT), r (reverse/OB/CTOB) |
| bwameth | YD |
f (forward/OT/CTOT), r (reverse/OB/CTOB) |
| gem3 / Blueprint | XB |
C (forward), G (reverse) |
These tags indicate which strand the original bisulfite/EM-seq-converted DNA came from, which is essential for correctly interpreting C/T as methylated/unmethylated.
Note: EM-seq (enzymatic methyl-seq) data produces the same C-to-T conversion pattern as bisulfite sequencing and is fully supported when aligned with any of the above tools.
-
Use the cache: The first run on a new genome builds a CpG site index, which is cached. Subsequent runs are much faster.
-
Process in parallel: bam2tensor processes one BAM at a time, but you can run multiple instances in parallel on different BAM files:
# Using GNU parallel find /data/bams -name "*.bam" | parallel -j 4 \ bam2tensor --input-path {} --reference-fasta ref.fa --genome-name hg38
-
Ensure BAM files are indexed: Each BAM file requires a corresponding
.bam.baiindex file. Create with:samtools index sample.bam
-
Use SSDs: Both reading BAM files and writing output benefit from fast storage.
-
Memory considerations: Memory usage scales with the number of CpG sites (columns) rather than reads. For human genomes (~28M CpG sites), expect moderate memory usage.
Main class for managing CpG site positions and coordinate conversions.
GenomeMethylationEmbedding(
genome_name: str, # Identifier for caching
expected_chromosomes: list, # List of chromosome names to process
fasta_source: str, # Path to reference FASTA
skip_cache: bool = False, # Force regeneration of cache
verbose: bool = False # Enable verbose output
)Key Methods:
embedding_to_genomic_position(embedding: int) -> tuple[str, int]- Convert column index to (chromosome, position)genomic_position_to_embedding(chrom: str, pos: int) -> int- Convert genomic position to column index
Key Attributes:
total_cpg_sites: int- Total number of CpG sites across all chromosomescpg_sites_dict: dict[str, list[int]]- Dictionary mapping chromosome names to lists of CpG positions
Core function for extracting methylation data from a BAM file.
extract_methylation_data_from_bam(
input_bam: str, # Path to BAM file
genome_methylation_embedding: GenomeMethylationEmbedding, # Embedding object
quality_limit: int = 20, # Minimum MAPQ
verbose: bool = False, # Enable verbose output
debug: bool = False # Enable debug output
) -> scipy.sparse.coo_matrixReturns: A SciPy COO sparse matrix with shape (n_reads, n_cpg_sites).
Contributions are welcome! Please see the Contributor Guide for guidelines on:
- Setting up a development environment
- Running tests
- Code style requirements
- Submitting pull requests
# Install development dependencies
uv sync
# Run all checks (linting, type checking, tests)
nox
# Run specific checks
nox --session=tests # Run pytest
nox --session=mypy # Type checking
nox --session=pre-commit # Linting
# Format code
uv run black src tests
uv run ruff check --fix src testsThis project includes a CLAUDE.md file with detailed guidance for AI coding agents (Claude Code, Copilot, Cursor, etc.). If you're using an AI assistant to contribute, please ensure it follows the project conventions described there.
Key points for AI agents:
- Run
noxbefore committing to validate all checks pass - Follow Google-style docstrings validated by darglint (long strictness)
- Use
uv syncfor dependency management - See
CLAUDE.mdfor complete guidelines
Distributed under the terms of the MIT license, bam2tensor is free and open source software.
If you encounter any problems, please file an issue with:
- A description of the problem
- Steps to reproduce
- Your Python version and operating system
- Relevant error messages or logs
Created and maintained by Nick Semenkovich (@semenko), as part of the Medical College of Wisconsin's Data Science Institute.
This project was generated from Statistics Norway's SSB PyPI Template.